マッピング
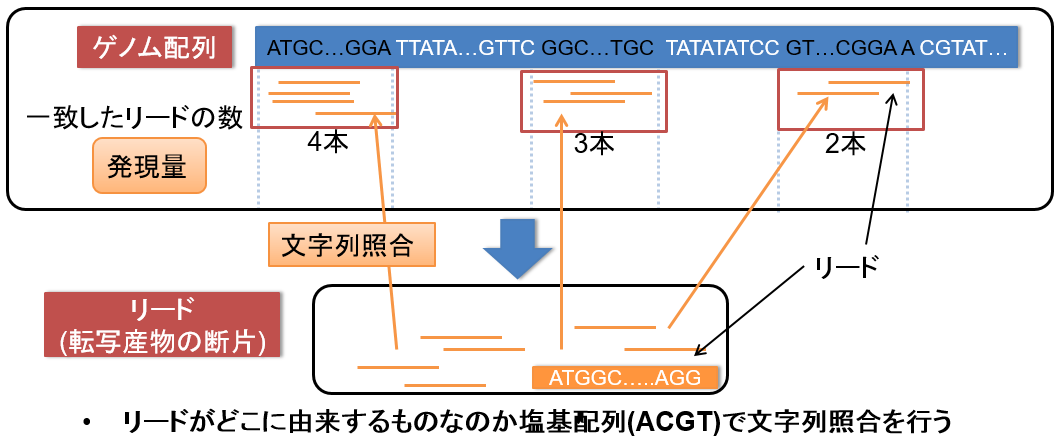
体内で起こっている現象を解明する手がかりとして,遺伝子がタンパク質として合成される量(発現量)というものがあります. 発現量を測定するためには,どの遺伝子がどれほどの量合成されているかという2つのことを調べる必要があります. しかし,これらのことをタンパク質について直接調べることは困難であるため,RNAという物質を用いて調べます. RNAは遺伝子からタンパク質が合成される際に生成される物質であり, このRNAから元になった遺伝子の塩基配列を復元することができます. 復元された塩基配列とゲノムを照らし合わせ,配列が一致する場所を調べることによって どの遺伝子が合成されているかという情報を調べることができます. そして,配列が一致するRNAの数を調べることによって合成されたタンパク質の量を推定します.
このように,発現量を測定するにはRNAから復元された塩基配列とゲノムを調べる処理が必要となります. この処理のことをマッピング(もしくはアラインメント)と呼び, 配列チームではより効率的で正確なマッピング方法を研究しています.
メタゲノム解析

今までの微生物の研究では,実験室で培養が可能な微生物を主な対象としてきました. しかしこのような微生物は少数派であり,ほとんどの微生物は実験室で培養できません. そこで近年,自然に生息する微生物を採取し研究する方法も取られています. この方法では培養できない微生物を研究することができるのですが, 自然から採取するため研究対象以外の微生物も試料に混入することになります. このように複数の生物が混じった試料を使うゲノム解析を行うことをメタゲノム解析といいます.
メタゲノム解析の第一歩としてメタゲノムから読み取った塩基配列(リード)が どの微生物のものかを調べる必要があります. これには既知のゲノムを学習させた単純ベイズ分類器を用いて行われています. しかし,従来の方法では学習データであるゲノムのサイズに大きく差があったため, 一部のリードは誤って分類されていました. 本研究では,ゲノム配列を複数に分割した後に学習を行うことによって分類精度を向上させました.